Submitting
The following classes must be submitted in order to be graded:
Make sure when you submit, all of the following are present and functional:
Class: LinkStrand
Constructor: LinkStrand()
Constructor: LinkStrand(String)
Method: public String toString()
Method: public IDnaStrand append(String)
Method: public IDnaStrand append(IDnaStrand)
Method: public long size()
Method: public IDnaStrand reverse()
Method: public ArrayList nodeList()
Method: public IDnaStrand cutAndSplice(String, String)
Method: public String getStats()
Method: public String strandInfo()
Method: public void initializeFrom(String)
You also must submit your README in order to receive any credit.
If you create any new classes which are required for the above classes to compile, you should submit those as well.
Linked List Overview
You’ll be creating a class LinkStrand that implements a Java interface IDnaStrand. The class simulates cutting a strand of DNA with a restriction enzyme and appending/splicing-in a new strand.
You must use a linked-list to support the operations — specifically the class LinkStrand should maintain pointers to a linked list used to represent a DNA strand. You should maintain a pointer to the first Node of the linked list and to the last Node of the linked list. These pointers are maintained as class invariants. A class invariant is a property that must hold true after the constructor is finished and at the entry and exit of all public member functions. In this case the property of pointing to first/last nodes must hold after any method in the class executes (and thus before any method in the class executes). A Strand of DNA is initially represented by a linked list with one Node; the Node stores one string representing the entire strand of DNA. Thus, initially the instance variables myFirst and myLast will point to the same node. If any nodes are appended, the value of myLast must be updated to ensure that it correctly points to the last node of the new list.
You’ll need a nested/inner class to represent a Node of
the linked list for storing DNA information. Here is what that should look like:
//Outer class definition....
public class Node {
String info;
Node next;
Node(String s){
info = s;
next = null;
}
}
private Node myFirst, myLast; //first and last nodes of list
private long mySize; //# nucleotides in DNA
...rest of class
Since the purpose of this assignment is to utilize Linked Lists, your LinkStrand should not use, nor should you need any global variables besides Nodes and primitive types. Using an unnecessary global data structure, such as an array, a StringBuilder, or an ArrayList, may cost you almost all or all of your correctness and engineering points.
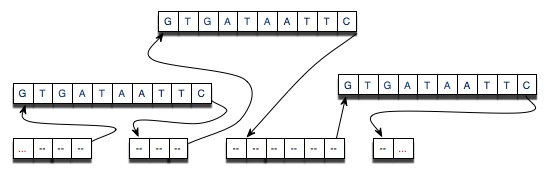
The diagram below shows the results of cutting an original strand of DNA (represented by "- - - ...")at three points and then splicing-in the strand “GTGATAATTC” at each of the locations at which the original strand was cut. Since splicing into a linked list is a constant-time, O(1) operation this implementation should be more efficient in time and space when compared to the String implementation.
How to Test Your Code
To test your LinkStrand class a JUnit tester has been provided for you. The tester will test individual methods in your class. If you need a refresher on JUnit refer to Discussion 4 Slides and Code.
Note: Passing these tests doesn't guarantee full credit since the tests are about correctness, not efficiency.
Implementing LinkStrand is the bulk of the coding work for this assignment. You’ll need to implement every method and use the JUnit tests to help determine if your methods are correctly implemented.
Overview of LinkStrand Methods
Because LinkStrand implements the IDnaStrand interface, all the methods you are overriding are documented with comments, so you should check there if you are wondering how any methods should work. You can also refer to the working methods in SimpleStrand, although they will differ in implementation because they use a StringBuilder instead of a linked list. Make sure you correctly implement every method specified in the interface. Below are some considerations for you as you begin to code:
-
Constructor and initializeFrom:
- Implement both with the same code (either copy the code or re-use the common code in one method)
- When creating/initializing a new LinkStrand only one node should be created. This node represents the entire string representation of DNA.
- Be sure to initialize the length of the simulated strand.
- Note: DNA Benchmark requires LinkStrand to have an empty constructor, i.e.
public LinkStrand(){ this(""); }
-
append:
- Implementation should not concatenate strings, but instead create new nodes
- If you’re appending a LinkStrand you should do so by redirecting pointers; you should not convert the strand to a string. You should be able to add any LinkStrand in O(1) time.
- If you're appending another type of Strand, (such as a SimpleStrand), you should convert the strand to a string and then append it, you can refer to the implementation in Simple Strand. (Hint: the instanceof operator will be helpful)
-
toString:
- Remember, your only class variables should be Node pointers and primitives. This means you should not use a global StringBuilder/String to keep track of the toString value (you can still use StringBuilders/Strings within the individual methods).
- Because of the way String addition works, combining many long Strings using a StringBuilder is faster than using +=.
-
cutandSplice:
- This method is NOT a mutator - the LinkStrand cutAndSplice is called from should be unchanged, and the method should be returning a new instance of LinkStrand.
- You may assume there is only one node (though it may contain a huge String of DNA). If there is more than one node throw an exception, e.g.
if(myFirst.next != null)) {
throw new RuntimeException("link strand has more than one node");
} - The implementation of this method should be virtually identical to the code in SimpleStrand except you'll be creating LinkStrand objects and calling LinkStrand.append to create the recombinant strand.
- Virtually identical means you simply replace SimpleStrand objects with LinkStrand objects and then the code should work. (Refer to the code in SimpleStrand to help you.)
- The diagram below gives an indication as to why the operation of splicing in a new strand will be O(B) for cutting at B places. The string splicee is the same for all of the Nodes being spliced in. Creating the node is therefore O(1) because the String splicee is already created.
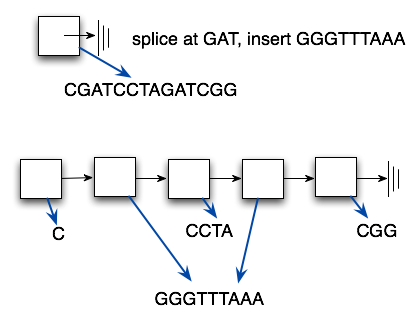
-
reverse:
- The SimpleStrand class uses the StringBuilder.reverse() method to reverse the simulated strand – note that this method is a mutator - it changes the StringBuider object it’s called on.
- In your LinkStrand class you could call .toString(), create a StringBuilder, and use .reverse() to create a single node LinkStrand object, but this will not earn you full credit. For full credit an N-node list should be reversed with an N-node list.
- For extra credit you should only reverse each unique string in the list once. This means that if a string/simulated strand of DNA has been spliced into a LinkStrand object the string should only be reversed/represented once.
-
size:
- Once the string is initialized, the length of a LinkStrand object should be calculated in O(1) time. Consider how you change the size of a strand in other methods.
-
getStats:
- Make sure this method displays the number of appends like in SimpleStrand. Just do that. Hint: You can refer to SimpleStrand to see how to implement getStats.
-
nodeList:
- The returned ArrayList should be in the same order - i.e. strand.nodeList().get(0) should be the same as the value in the head of your linked list.
- The return value for this should be generated at the time the method is called (since you are not allowed to maintain a global ArrayList).
Benchmarking Part 2
We're going to see how our newly created LinkStrand fares in the benchmark.
Let's run virtual experiments to show that LinkStrand is O(B) where B is the number of breaks. First, change SimpleStrand to LinkStrand in the main method of the class DNABenchMark. Then, make different runs to demonstrate this O(B) behavior, changing the number of breaks in your file. You can do this by constructing your own genomic data or by reusing ecoli.dat.
In your Analysis, describe the process, the results, and the reasoning you used to conclude the code is O(B). Please write down all the data you gathered, including timings, that demonstrate this O(B) behavior; graph the data to strengthen your case, and explain your data by demonstrating you understand the process.
Analysis
- Data, explanation of process, and explanation of results for
SimpleStrand.cutAndSplice. In particular, you should generate data to display O(N) behaviour where N is the size of the recombined strand returned. For this, just run the DNABenchmark as given to you and put your data into README. Explain why O(N) makes sense. Record your results in Analysis. - Data, explanation of process, and explanation of results for longest string and time for both 512M and 1024M heap-size. In your analysis, comment on whether you can fit in the next power-of-two string into 1024M after 512M and comment on the timing. (Did the time increase for the last entry? If so, by how much? You can change the memory size to 2048M and 4096M to do an even better analysis) Describe how you determined the power-of-two string you can use in both memory sizes, what it is, and the amount of time it took. For this section, the value which we'll care about is when the program crashes due to the heap space error.
- Data, explanation of process, and explanation of results for the virtual experiments ran for LinkStrand, including your conclusion and explanation of O(B) time complexity where B is the number of breaks. The instructions for this step is given clearly and concisely in Benchmarking Part 2.
Your analysis should contain:
Grading
For transparency’s sake, below is a list of aspects of your code the automated tests will check. Your code will still be checked by a TA, and passing all these tests does not guarantee a perfect grade.
FAQ
Here are a few commonly asked questions. This will be updated through the semester and through the years.
###Q: Can I have an example of an enzyme and a splicee?

###Q: When implementing my LinkStrand class and then running it through the DnaBenchmark, two of my recomb values returned as negative numbers. Any ideas why that might happen?
###A: Sounds like an integer overflow, your two largest recomb strings are probably longer in length than int’s max value, and if you try to store a value above the max int value, it will instead overflow to the minimum int value (a large negative number) and continue counting from there. If you want to see the larger numbers, try using a long instead (the same as an int, but has more bits and thus can store larger values).
###Q: What should cutAndSplice return if the given enzyme cannot be found within the strand?
###A: An empty string.
###Q: How do I vary the number of breaks in proving LinkStrand cutAndSplice is O(B)?
###A: The easiest way to do this is to take the sample files we gave you and copy/paste them over and over into new text files. So, for example, say ecoli.txt has b breaks. Then, create ecoli2.txt, which has ecoli.txt pasted into it twice. Logically, it should have 2b breaks, since every instance of the enzyme has been pasted twice. Do the same for an ecoli3.txt and it has 3b breaks. This requires little effort, and creates nicely spaced out numbers of breaks, which will give you a more accurate regression.
If you are unable to create larger files due to memory issues, you can also use the same data file and change the enzyme. Alternatively, instead of copy/pasting all of ecoli.txt, copy/paste part of it.
Note that DNABenchmark removes non-acgt characters upon loading a new file.
###Q: My LinkStrand cutAndSplice looks like it works, but when I run TestStrand it returns the original strand and I get a ‘self alter fail’.
###A: Unlike every other method, in LinkStrand.cutAndSplice you should not modify/return this - instead, you should create a new instance of LinkStrand which is a copy of this, modify that, and return that instead. So if your original code for cut and splice looks like
methoda();
methodb();
return this;
You instead want
IDNAStrand ret = new LinkStrand(this.toString());
ret.methoda();
ret.methodb();
return ret;
Theory
Restriction Enzyme Cleaving
The science behind the DNA Assignment
Restriction enzymes cut a strand of DNA at a specific location, the binding site, typically separating the DNA strand into two pieces. In the real chemical process a strand can be split into several pieces at multiple binding sites, we’ll simulate this by repeatedly dividing a strand.
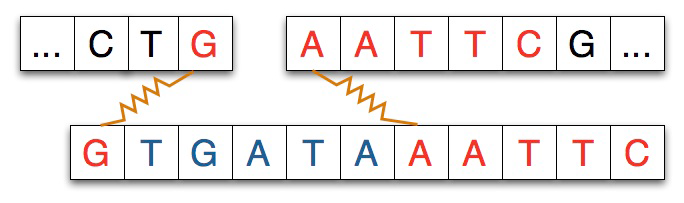
Given a strand of DNA aatccgaattcgtatc and a restriction enzyme like EcoRI gaattc, the restriction enzyme locates each occurrence of its pattern in the DNA strand and divides the strand into two pieces at that point, leaving either blunt or sticky ends as described below. In the simulation there’s no difference between a blunt and sticky end, and we’ll use a single strand of DNA in the simulation rather than the double-helix/double-strand that’s found in the physical/real process.
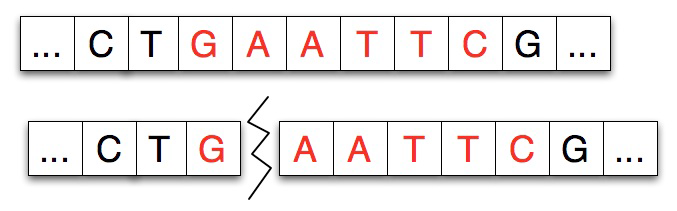
Restriction enzymes have two properties or features: the pattern of DNA that marks
a site at which separation occurs and a number/index that indicates how many
characters/nucleotides of the pattern attach to the left-part of the split strand. For
example, the adjacent diagram shows a strand split by EcoRI. The pattern for EcoRI
is gaattc and the index of the split is one indicating that the first
nucleotide/character of the restriction enzyme adheres to the left part of the split.
In some experiments, and in the simulation you’ll run, another strand of DNA will be
spliced into the separated strand. The strand spliced in matches the separated
strand at each end as shown in the diagram below where the spliced-in strand
matches with G on the left and AATTC on the right as you view the strands.